Researchers’ collaborative efforts from Denmark and Germany used BioMAX beamline to discover selective, non-covalent inhibitors of Keap1– a common target against oxidative stress and inflammation. The result is a potent antithesis of the currently available drugs acting as covalent inhibitors.
In improving oxidative stress and inflammation treatments, such as sclerosis and various kidney, respiratory, skin, and joint diseases, protein-protein interaction (PPI) between Nrf2 and Keap1 has become a key observation point. However, current commercial Keap1-targeting drugs covalently inhibit Keap1, a mechanism of action that comes with a lack of selectivity, side effects, and other complications during drug development.
“Through this fragment-based drug discovery (FBDD), we made non-covalent inhibitors, which are believed to be more selective and cleaner in their action. Therefore, our compounds can become a useful tool for further validation and scientific exploration of Keap1 as a drug target, and potentially the compounds can open the gate for drug discovery projects leading to better medicine and drugs for currently unmet needs,” said Principal Investigator Anders Bach from the University of Copenhagen.
Team’s first FBDD research with great results
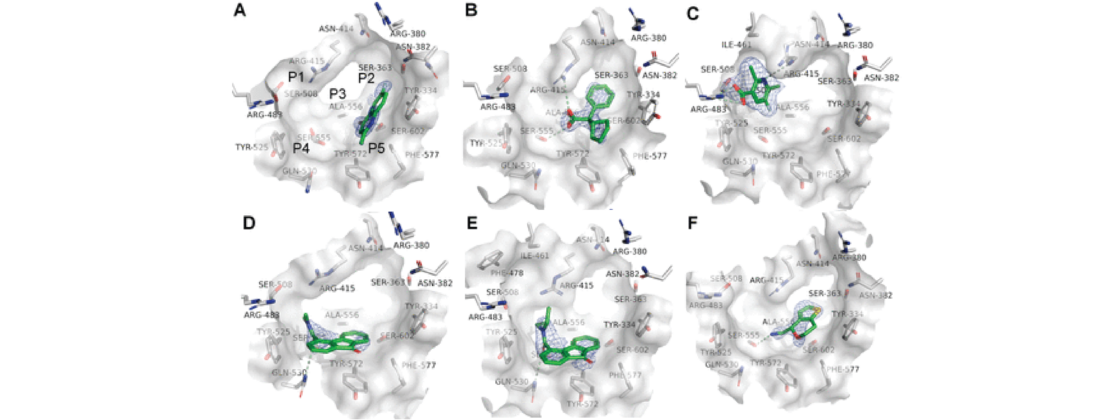
In this first FBDD research, scientists screened 2500 fragments using an array of techniques, including X-ray crystallography experiments done at ESRF, DESY, and MAX IV.
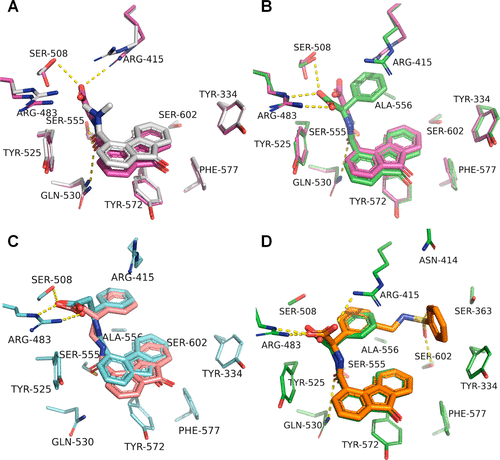
“We optimized one of the fragments to high-affinity non-covalent Keap1-Nrf2 inhibitors in this project. One of our biggest learning was that the fragments, and their analogues, can twist and turn in the binding pocket, and not all visually proven binding modes represent good starting points for structure-based drug design. Finding out which fragments were amenable to optimization took a lot of trial and error and was hard to rationalize,” said Bach.
Researchers implemented several screening and validation methods to pick the best hits as the initial SPR screen resulted in around 400 low-affinity hits. They also soaked many fragment hits and analogues into Keap1 crystals, which was pivotal in the research.
“To see these methods play together to provide highly well-characterized fragment hits was and still is awesome. The fact that we could convert a weak (about 500 µM) affinity fragment to a potent lead compound by medicinal chemistry and X-ray crystallography is remarkable. Overall, our fragment-to-lead (F2L) efforts led to a 1700-fold improvement in affinity. The work provides the foundation for our FBDD platform that we are now applying to other drug targets,” explained Bach.
The team now continues to develop Keap1 inhibitors. Their years of research have resulted in new high-affinity and drug-like compounds, which will be published soon.
“We plan to test the best compounds in animals soon for PK properties, and hopefully, this will lead to testing in animal disease models with key collaborators. We also consider innovation opportunities and have filed patents, as we think our compounds have the potential to become useful,” explained Bach.



